وبلاگ
تالاسمی و روش های تشخیص آزمایشگاهی
تالاسمی نوعی کم خونی از گروه بیماری های “هموگلوبینوپاتی” است که در اثر نقص در ژن های سازنده هموگلوبین ایجاد میشود. در اثر این بیماری هموگلوبین ساختار طبیعی خود را از دست میدهد و هموگلوبین غیر موثر ایجاد میشود. هموگلوبین های ناقص نمیتوانند به مقدار کافی به سلول ها اکسیژن برسانند. در واقع در این بیماری کمبود کلی هموگلوبین وجود ندارد بلکه مقدار هموگلوبین غیرطبیعی بیشتر از نوع طبیعی تولید میشود.
ساختار هموگلوبین
هموگلوبین، پروتئین اصلی گلبول قرمز است که وظیفه ی انتقال اکسیژن را انجام میدهد. ساختار این پروتئین شامل 4 زنجیره ی گلوبین میباشد که در هر زنجیره گلوبین یک مولکول هم وجود دارد. مولکول های هم دارای آهن می باشند که بوسیله ی آن اکسیژن را حمل میکنند.
اندازه طبیعی هموگلوبین خون در آقایان 2± 14 و در خانم ها 2± 12 گرم در دسیلیتر (g/dl) میباشد.
انواع هموگلوبین بر اساس نوع زنجیره:
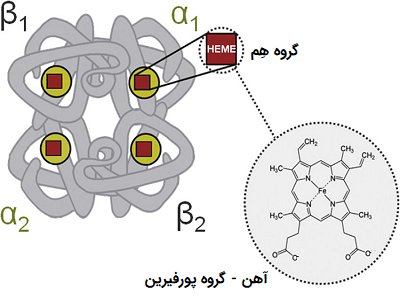
هموگلوبین A : این نوع هموگلوبین از دوزنجیره ی آلفا و دو زنجیره ی بتا تشکیل شده است که یک ساختار تترامر (4 تایی) را تشکیل میدهد. این ساختار α2β2 نامیده میشود. در انسان بالغ و سالم هموگلوبین A حدود 98% کل انواع هموگلوبین موجود در گردش خون را تشکیل میدهد.
هموگلوبینA2 : شامل 2 زنجیره آلفا و 2زنجیره دلتا میباشد (α2δ2). HbA2 بطور طبیعی 2-1% هموگلوبین در بالغین سالم را تشکیل میدهد.
هموگلوبین F : هموگلوبین اصلی دوران جنینی است. هموگلوبین F کمتر از 1% هموگلوبین در بالغین را نیز تشکیل میدهد. این نوع از دو زنجیره آلفا و دو زنجیره گاما (α2δ2) ساخته شده است.
هموگلوبین های C ، H و … نیز وجود دارند که در برخی بیماری ها بوجود میآیند.
انواع تالاسمی
ژن های سازنده ی زنجیره های هموگلوبین توسط هر دو والد به ارث میرسند. بر اساس جهش یا حذف در هر یک از این ژن ها ساخت زنجیره ی گلوبینی مربوطه مختل میشود و بر همین اساس انواع تالاسمی بوجود می آید.
تالاسمی به دو نوع آلفا و بتا تقسیم میشود. نوع بتا شامل تالاسمی ماژور و مینور میباشد.
آلفا تالاسمی:
در نوع آلفا تالاسمی، پروتئین آلفا هموگلوبین تولید نمی شود. این نوع از بیماری در مدیترانه، آفریقا، خاورمیانه، هند، آسیای جنوبی و جنوب چین شایع است.
۴ گونه ی مختلف تالاسمی آلفا وجود دارد که با توجه به عوارض آن ها از خفیف تا شدید تقسیم بندی شده است. این مراحل شامل: مرحله حامل خاموش، نوع Trait یا خفیف، هموگلوبین H و هیدروپس فتالیس یا آلفا تالاسمی ماژور می باشد.
بتا تالاسمی:
حذف یا جهش در یک یا هر دو ژن سازنده زنجیره بتا، منجر به ایجاد بتا تالاسمی میشود.
تالاسمی مینور
در این نوع کمبود زنجیره بتا به حدی نیست که باعث اختلال در عملکرد هموگلوبین شود. افراد مبتلا به تالاسمی مینور تنها ناقل ژنتیکی بیماری هستند و نیازی به درمان ندارند. این افراد به جز یک کم خونی خفیف در برخی موارد، مشکل دیگری را تجربه نخواهد کرد. تنها مواردی که در این بیماری به مراجعه به پزشک نیاز میشود ممکن است درمان کم خونی فقر آهن یا درمان تجویز نادرست مکمل آهن باشد.
تالاسمی بینابینی
در این حالت کمبود پروتئین بتا در هموگلوبین باعث کم خونی نسبتاً شدید میشود. اختلالات قابل توجهی در سلامت فرد مانند بدفرمی های استخوانی و بزرگی طحال در این نوع از بیماری دیده میشود. در این مرحله طیف وسیعی از علائم وجود دارد.
به دلیل نیاز این بیماران به تزریق خون، آن ها را در گروه تالاسمی ماژور قرار می دهند. بیماران مبتلا به تالاسمی بینابینی برای بهبود کیفیت زندگی و نه برای نجات یافتن، به تزریق خون نیازمندند.
تالاسمی ماژور با کم خونی Cooley’s Anemia

نوع ماژور شدیدترین فرم تالاسمی بتا می باشد که کمبود شدید پروتئین بتا در هموگلوبین منجر به یک کم خونی تهدیدکننده حیات می شود. افراد مبتلا به این بیماری به انتقال خون منظم و مراقبت های طبی فراوانی نیازدارند.
دفعات مکرر انتقال خون در طول عمر این افراد باعث تجمع بیش از حد آهن می شود. آهن اضافی در ارگان هایی مثل کلیه رسوب میکند و میتواند باعث نارسایی و مرگ ارگان ها شود. برای جلوگیری از این اتفاق، آهن اضافی باید توسط تجویز داروهای شلاته کننده ی آهن (Chelator) دفع گردد.
تشخیص آزمایشگاهی
آزمایش خون CBC برای تشخیص اولیه تالاسمی در فرد تجویز میشود. در این آزمایش پارامتر های تعداد سلول های خونی (RBC, WBC, Plt)، اندازه، حجم گلبول های فرمز (MCV, HCT,RDW) مقدار و غلظت هموگلوبین (HGB, MCH, MCHC) اندازه گیری میشود. تمام این پارامتر ها بطور اتوماتیک توسط دستگاه سل کانتر اندازه گیری شده و لام گسترش خون نیز بصورت دستی بررسی میشود.
همچنین انجام آزمایش الکتروفورز هموگلوبین، معاینه بالینی، سونوگرافی طحال به تشخیص تالاسمی کمک میکنند. و در صورت ناقل یا مشکوک بودن فرد به تالاسمی، به مراکز ژنتیک معرفی می گردد.
آزمایش های ژنتیکی شامل بررسی DNA و تشخیص فرد حامل ژن های معیوب هموگلوبین با استفاده از تکنیک های تخصصی MLPA , Sequencing , ARMS PCR و یا انجام آزمایشات جنینی در هفته های اول بارداری میباشد.
نوع مینور این بیماری را با سنجش انداره گلبول های قرمز و مقدار هموگولبین A2 تشخیص می دهند. پس قبل از ازدواج و فرزند آوری لازم است حتما آزمایش مربوطه را انجام دهید.
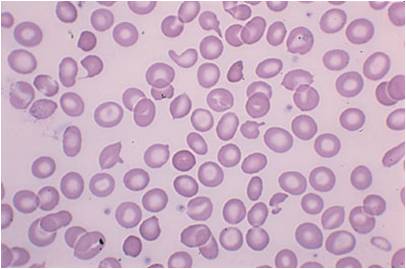
در نوع ماژور مقدار هموگلوبین در آزمایش CBC به کمتر از ۵ گرم در دسی لیتر می رسد.
همچنین شکل و رنگ گلبول های قرمزغیر نرمال می شود. با استفاده از میکروسکوپ می توان شکل و کم رنگ شدن گلبول های قرمز را تشخیص داد (میکروسیت-هیپوکروم) .
شرایط انجام آزمایش خون
نیاز یه ناشتائی نمیباشد. در صورت مصرف مکمل حاوی آهن، باید به پزشک معالج اطلاع داده شود. نمونه ی مورد بررسی نمونه خون وریدی می باشد ولی در صورت درخواست بررسی وضعیت جنین از لحاظ ابتلا به تالاسمی ماژور ، نمونه از پرزهای جنینی یا 20 میلی لیتر از مایع آمنیوتیک جنینی (آمنیوسنتز) برداشته میشود.
علائم بیماری
افراد براساس وخامت بیماری دچار کم خونی، سرگیجه، خستگی مفرط، ضعف و بیحالی شدید، افزایش حجم مغز استخوان صورت و جمجمه، اختلال رشد، بزرگی کبد و طحال، افزایش غیر طبیعی آهن (Iron overload)، مشکلات قلبی و… میشوند.
تالاسمی چگونه به ارث میرسد؟
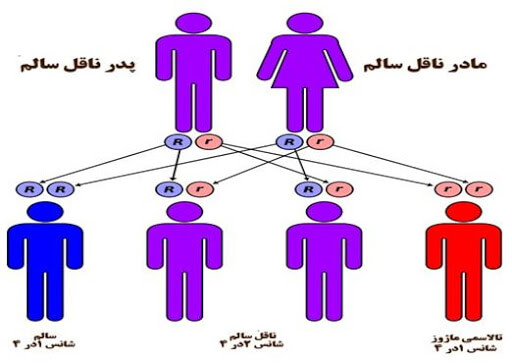
تالاسمی جزو بیماری های اتوزومال مغلوب میباشد. والدینی که هر دو به بیماری تالاسمی مینور مبتلا هستند احتمال دارد نوزادانی با بیماری تالاسمی ماژور به دنیا بیاورند. ممکن است این افراد خود هیچ علامتی برای این بیماری نداشته باشند ولی برای پیشگیری از تولد نوزادی با تالاسمی ماژور باید آزمایش خون تالاسمی انجام شود.
با توجه به درمان سخت این بیماری که شامل تزریق خون در هر ماه و تا آخر عمر و مصرف داروی آهن می باشد، بهترین راه حل برای این مسئله پیشگیری و آگاه سازی افراد می باشد.
غربالگری بیماری
آزمایش خون تالاسمی در این افراد مورد نیاز است:
- افراد مبتلا با علائم مشابه آلفا تالاسمی.
- افراد خانواده فرد مبتلا که احتمال دارد ناقل باشند.
- برای تشخیص بیماری تالاسمی در جنین در صورتی که نوع جهش والدین از قبل مشخص شده باشد.
- زوجین ناقل (مینور) (نمونه خون والدین زوجین نیز برای تعیین پیوستگی لازم است).
پیشنهاد بررسی: لوله وکیوم CBC K2, K3 لبویر – لوله لخته کلات بدون ژل(Clot Activator) لبویر – لوله وکیوم لخته ژل دار لبویر